THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
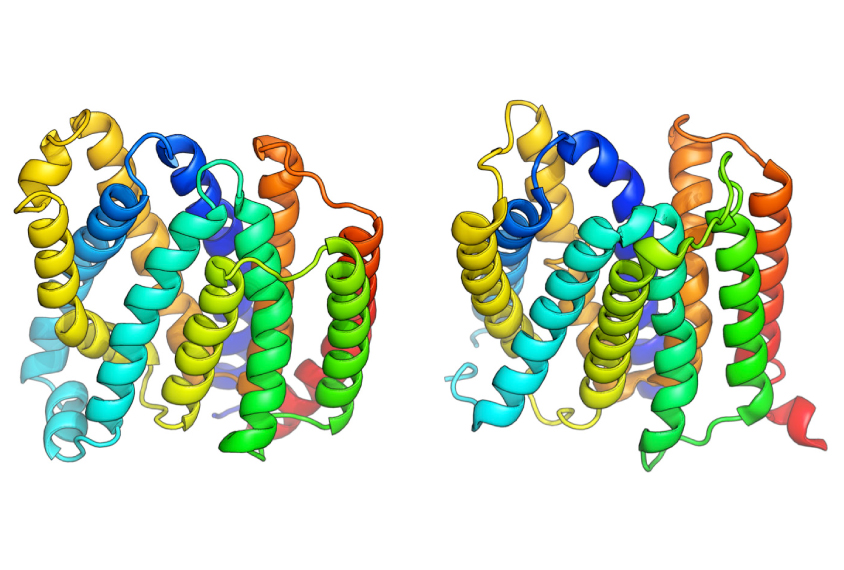
A new computer program can predict the 3-D structure of proteins at a fraction of the cost of traditional methods1. It revealed the configuration of more than 600 proteins of distinct types that have been difficult to characterize using standard techniques.
A protein is a chain of molecules called amino acids. That chain folds into a 3-D structure dictated by the sequence of amino acids. This structure determines the protein’s function.
There are roughly 15,000 known protein families, each of which typically contains a thousand or more similar proteins. Techniques such as X-ray crystallography and nuclear magnetic resonance spectroscopy have revealed the structures of proteins in about a third of these families. Computer models have determined the shapes in nearly an additional third based on their similarity to known proteins.
The remaining proteins lack cousins with known structures, so scientists must find a way to predict each protein’s structure from its amino acid sequence.
A computer program called Rosetta can generate all the possible structures of a particular protein based on the protein’s amino acid sequence. It can then calculate which of those conformations are the most stable and biologically plausible. But the program requires a lot of computational power. And a protein can have several stable conformations, making it difficult to know which one exists in nature.
A new version of Rosetta, described 19 January in Science, narrows down the possible structures of a particular protein by identifying amino acids that typically come together when proteins fold. The program compares gene sequences for similar proteins in large numbers of microbes. It then picks out amino acid pairs that evolution favors and that likely serve as critical ‘joints’ for proteins.
Using the upgraded program, researchers generated the structures for representative proteins from 614 elusive protein families. X-ray crystallography has confirmed five of Rosetta’s predictions.
By joining the discussion, you agree to our privacy policy.