THIS ARTICLE IS MORE THAN FIVE YEARS OLD
This article is more than five years old. Autism research — and science in general — is constantly evolving, so older articles may contain information or theories that have been reevaluated since their original publication date.
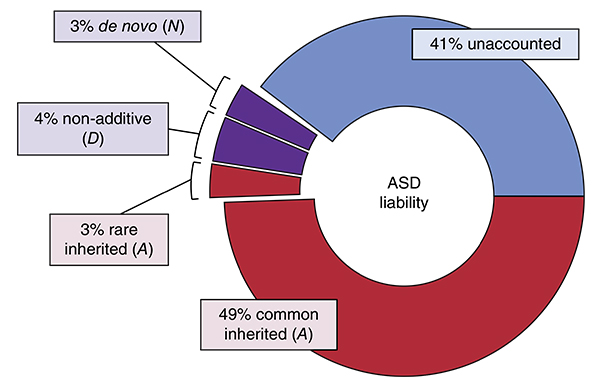
Genetic risk: Using multiple statistical models, researchers have attributed 59 percent of the risk of developing autism to genetics.
Common gene variants that have minor effects may contribute about half the risk of developing autism, according to a study published Sunday in Nature Genetics1. Identifying these variants would require tens of thousands of samples.
Much of autism research so far has focused on rare, de novo mutations, which appear spontaneously in individuals with autism. These mutations often have strong effects and so can be straightforward to find — for example, by comparing the genomes of the affected individuals with those of their unaffected family members.
By contrast, common variants are present in 5 percent or more of the population. Alone, each variant may have little effect on an individual, but taken together they can tip the scales toward a particular condition, such as autism.
Because of their prevalence, definitively linking a particular variant to a disorder requires tens of thousands of samples.
Rather than pinpoint individual variants, the new study looked at how much common variants contribute to autism risk overall. The results suggest that roughly 49 percent of the risk of developing autism can be attributed to common variants, versus 3 percent for rare, de novo variants.
“De novo mutations are extraordinarily important, but we need to consider this other kind of inherited risk as a critical part of the [genetic] architecture,” says lead researcher Joseph Buxbaum, director of the Seaver Autism Center at the Icahn School of Medicine at Mount Sinai in New York City.
Family ties:
Several studies have tried to define exactly how much of autism risk can be attributed to inheritance. These studies often analyze the genomes of twins because identical twins share the same DNA. However, they don’t always adequately account for the fact that twins also share other factors — such as the in utero environment, their homes or pediatricians.
“In the world of heritability, which is something of a dark art, twin studies are the darkest of the arts,” says Stephan Sanders, assistant professor of psychiatry at the University of California, San Francisco.
Perhaps as a result, estimates from twin studies of genetics’ contribution vary widely — ranging from 90 percent in twin studies in the 1980s to only 37 percent in a controversial 2011 twin study.
The new study falls in the middle, estimating the contribution of genetics overall at around 59 percent.
The researchers looked at autism risk across all children born in Sweden between 1982 and 2007, including 5,689 diagnosed with autism, in a total of 1.6 million families.
The researchers pieced together pedigrees of extended families to an unprecedented level, from first-degree relatives (parents, siblings) to ninth-degree distant cousins. By looking at autism recurrence across these families, they calculated that about 52 percent of autism risk is inherited.
In the world of heritability, which is something of a dark art, twin studies are the darkest of the arts.
The “magic” of this analysis is that the distant relatives — many of whom don’t even know each other — are less likely to share a home or other environmental confounds than siblings are, says Kathryn Roeder, professor of statistics at Carnegie Mellon University in Pittsburgh.
To identify the role of common variants, the researchers looked at more than 500,000 variants shared among 3,046 unrelated individuals in the population. From this, they estimated that common variants contribute about 49 percent of autism risk overall. That suggests that only the remaining 3 percent of risk comes from rare, inherited mutations.
These calculations are estimates, with sizable error rates. A study of Swedish twins published earlier this month similarly pegged the risk from inherited genetics at 54 percent, however, boosting the numbers’ credibility2.
“By using a variety of different analytical approaches, the researchers still come up with the same figure of around 54 percent,” says Louise Gallagher, professor of child and adolescent psychiatry at Trinity College, Dublin, who was not involved in the study. “The strength [of the new study] is the large population sample.”
Spontaneous risk:
The new study also calculated the contribution of de novo mutations — which are not included in estimates of inherited risk — at about 3 percent. This calculation may be an underestimate, say experts. For example, the analysis includes only mutations found in the coding portion of the genome.
Still, the relatively low contribution from de novo variants belies the importance the field has given to them so far, says Dan Arking, associate professor at the Institute of Genetic Medicine at John Hopkins University in Baltimore, who was not involved in the study. “This is bringing people back to the reality that common variation is explaining most of autism risk,” he says.
Together, all these genetic factors (along with a previously published estimate of recessive mutations) add up to 59 percent of autism risk.
Although other studies have designated the remainder as ‘environmental’ risk, the researchers say this category should be considered “unaccounted.” This is because the analysis does not account for interactions between risk factors — between common and rare variants, for example, or the influence of environment on gene expression.
De novo mutations may in fact act as a ‘second hit’ that pushes a set of common variants toward autism, says Buxbaum.
“The inter-relationship between common and rare variation and inherited and de novo variation is going to be the big thing for the next few years,” he says.
In any case, it’s clear that large numbers of samples can transform autism research — as they have in work on schizophrenia.
Just five years ago, schizophrenia researchers had access to only a few thousand genomes — enough to show that common variants are important in the disorder, but not enough to identify any particular variant — says Benjamin Neale, assistant professor of analytic and translational genetics at Massachusetts General Hospital.
But after analyzing nearly 40,000 schizophrenia genomes, an international consortium of researchers identified more than 100 common variants associated with schizophrenia — 83 of which are new links to the disorder.The researchers published their results yesterday in Nature3.
“The kind of trajectory we’ve seen in schizophrenia suggests what the future of autism genetics may hold — if we make a strong commitment as a community to increasing sample sizes,” says Neale.
References:
1: Gaugler T. et al. Nat. Genetics Epub ahead of print (2014) PubMed
2: Sandin S. et al. JAMA 311, 1770-1777 (2014) PubMed
3: Schizophrenia Working Group of the Psychiatric Genomics Consortium. Nature Epub ahead of print (2014) Abstract
By joining the discussion, you agree to our privacy policy.